Origami of the Cell


In the ancient Japanese art of origami, paper must be folded precisely and following a specific order to create the desired result — say, a crane or lotus flower. It’s a complex pursuit that requires keen attention to detail and utmost accuracy.
An equally precise biological process in living cells gives rise to proteins, the large biomolecules essential for life.
Proteins begin life as long strings of amino acids that must fold into the three-dimensional shape prescribed for their particular biological function. When proteins don’t fold as expected — think badly misshapen crane — the cells activate stress responses meant to mitigate the problem. But severe or prolonged stress produces an acute response: Cell death is triggered to protect the organism.
Sustained activation of one such reaction — the unfolded protein response, or UPR — has been implicated in a number of diseases. Seeking to illuminate a piece of this biological puzzle, an international team of scientists, including UC Santa Barbara cell biologist Diego Acosta-Alvear, examined the role of a central UPR component, a stress sensor protein called IRE1 (inositol-requiring enzyme 1), in atherosclerosis.
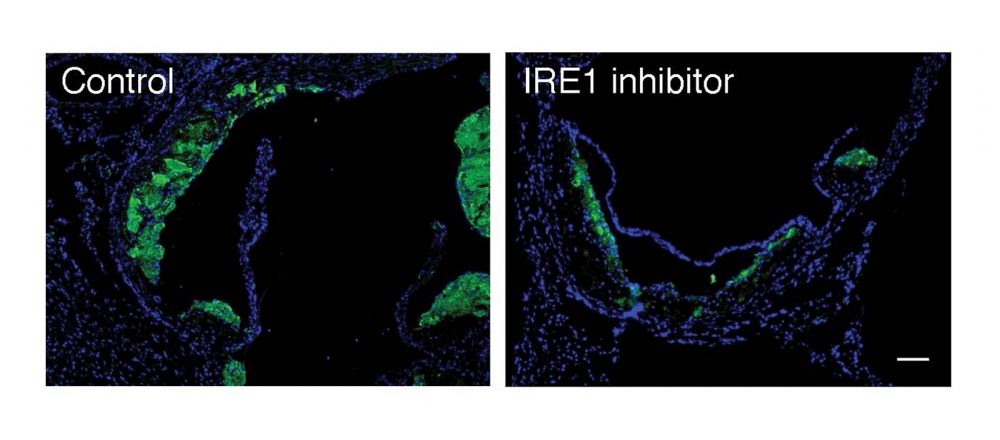
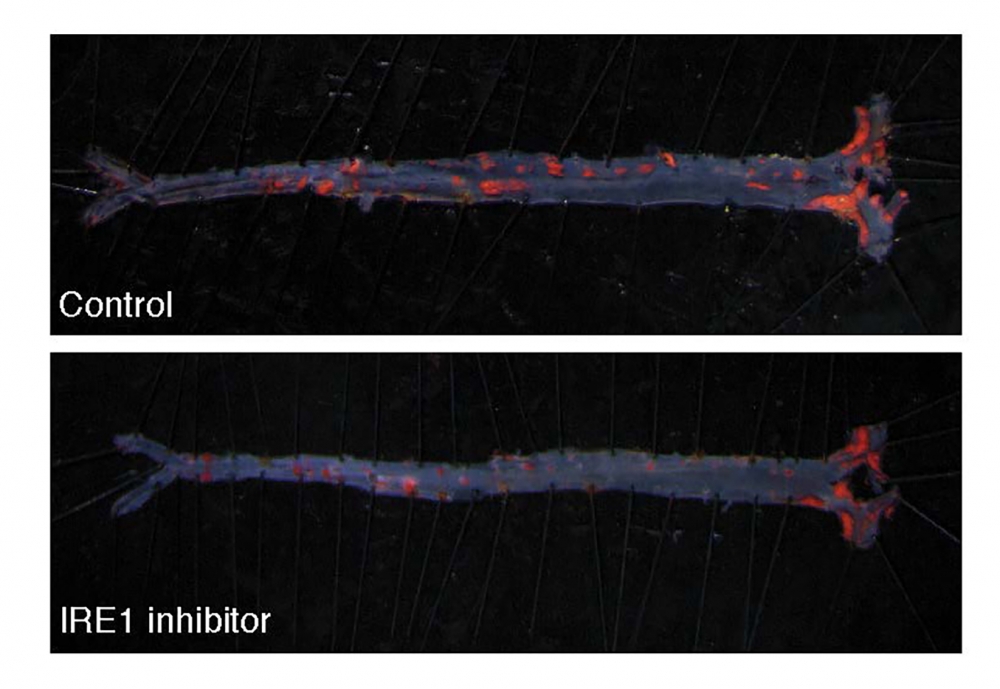
The researchers found that blocking IRE1 with a small molecule prevented the progression of atherosclerosis in mice. The findings appear in the Proceedings of the National Academy of Sciences.
“A healthy cell has one type of stress response network wiring and it’s likely that a diseased cell accommodates that wiring to survive,” said Acosta-Alvear, an assistant professor in the Department of Molecular, Cellular and Developmental Biology. “Stress response networks control the life vs. death decision in cells, and since a diseased cell is nowhere near its comfort zone, rewiring its stress responses allows it to avoid or delay cell death even when conditions are adverse. That’s what we wanted to understand: how a diseased cell does that and why it happens.”
The UPR is triggered when the normal functions of the endoplasmic reticulum — the cell’s largest organelle in charge of making and folding proteins — are compromised. Though the UPR usually promotes healthy endoplasmic reticulum function, sustained UPR activation sometimes results in diseases such as atherosclerosis, the deposition of fatty plaques on artery walls, among other conditions. Understanding what happens with the UPR in disease is key to illuminating the normal operation of this essential pathway — and to providing insights into the development of targeted therapies.
Endoplasmic reticulum stress is triggered not only by protein-folding problems, but also by fatty acids, explained Acosta-Alvear. Fat-induced stress and metabolic overload of the endoplasmic reticulum can alter its function, triggering chronic inflammation, which plays an important role in the development of atherosclerosis.
In this research, the scientists disturbed endoplasmic reticulum function by introducing saturated fatty acids into cells to induce lipotoxic stress. This in turn activated the UPR and IRE1.
Active IRE1 relays the protein-folding stress information to the cell nucleus by controlling the production of a very potent transcription activator, XBP1 (X-box binding protein-1). Transcription activators are proteins involved in the process of converting, or transcribing, DNA into RNA.
The investigators’ analyses demonstrated that XBP1 was responsible for turning on pro-atherogenic genes. They then treated mice with a compound that blocked IRE1.
“The end result was that if the transcription factor was not produced, the pro-atherogenic genes were not turned on, which mitigated the progression of the disease,” Acosta-Alvear said. “This research is a proof-of-concept study showing that blocking this single critical enzyme delivers a desirable therapeutic benefit. It’s a first step in mechanistically understanding how cellular stress responses are wired in specific contexts.”
Share this article
Related Stories



