
Researchers at UC Santa Barbara have discovered a molecular pathway that may explain how a particularly deadly form of cancer develops. The discovery may lead to new cancer therapies that reprogram cells instead of killing them. The findings are published in a recent paper in the Journal of Biological Chemistry.
The UCSB research team described how a certain mutation in DNA disrupts cellular function in patients with acute myeloid leukemia (AML). The researchers were prompted to study this process by another research team's discovery that AML patients have a mutation in a certain enzyme, which was reported in the New England Journal of Medicine. The enzyme is a protein called DNMT3A, which leads to changes in how the DNA of AML patients is methylated, or "tagged." Norbert Reich, professor in the Department of Chemistry and Biochemistry at UCSB, was already studying that particular enzyme with his research group, so they began to study the disease process of AML at the cellular level.
Reich explained that tagging is a way of reading DNA at the cellular level. This falls within an area of study called epigenetics, a process that occurs "on top" of genetics. Each person has approximately 200 types of cells, all with the same DNA, and these must be controlled in different ways. "There is an enzyme –– a protein –– that tags DNA and controls which of the genes in your cells, your DNA, gets turned on and off," said Reich. "So you have 20,000 genes, and you have to control them differently in your brain than in your liver."
Reich explained that there is current interest in this broader field of epigenetics as a direction for the treatment of cancer. "There's definitely the idea that this may be a new way of developing therapeutics, because you don't have to kill the cancer cell," said Reich. "Almost every cancer therapy that's out there works on the principle that a cancer cell needs to be killed."
With epigenetics, instead of only having DNA sequence coding for certain genes, there is an epigenetic process, with another layer of information on top of the genetic process. In this case, that information is the tagging by the methyl groups.
"If you really think about it, this is part of the answer as to how your cells can be so different and yet they all have the same DNA," said Reich. "You have the same genome in every one of your cells, but you do not have the same epigenome, which is basically the methylation pattern, the tagging pattern. That is different in every type of your cells. And the way this relates back to cancer, with leukemia, in those patients, the tagging is messed up. The patterns are not correct. Our big contribution to that is we've explained how the mutations in the enzyme could lead to that disruption of the tagging pattern."
The UCSB group developed a test to demonstrate that the mutant enzymes in AML can only work on DNA for short distances. As a result, the precise methylation patterns of a healthy cell are disturbed, resulting in genes being turned on at the wrong place and time, which in turn can initiate the growth of cancerous cells.
The team found that the mutation AML patients have causes a certain complex of four proteins to be disrupted. "The surprise was that the disruption doesn't stop the enzyme from being active; it doesn't stop the enzyme from tagging the DNA," said Reich. "Instead, it stops the way it can do it. Instead of going to your DNA and tagging an entire region of chromosome, it goes there, does one thing, and leaves. That process, that change, is what we see in the AML patients. So we think we have a molecular explanation for this disease."
Reich said that the currently prescribed drug Vidaza works by affecting the same enzyme that is mutated in AML. There is interest in the pharmaceutical industry in developing other therapeutics to target the enzymes responsible for tagging the DNA. These epigenetic inhibitors would reprogram rather than kill the cell.
Traditional cancer therapies use radiation and chemotherapy to remove or kill cancer cells. "The problem with that is that cancer cells are often very subtly different from normal cells," said Reich. "So you have one of the most difficult therapeutic challenges known to man, which is to distinguish between two human cells –– one that's cancerous and one that's not. Instead of killing the cell, the notion is that if you could just reprogram the cell, then it goes back to being normal. You intercept the cancer development. This is still an aspiration; it hasn't been achieved really, but that's what attracts people to the field of epigenetic-based therapies, because of the prospect of not having to kill cells."
Celeste Holz-Schietinger and Douglas Matje, both graduate students working in the Reich lab, are the first and second authors of the paper.
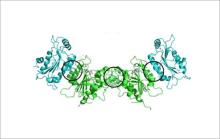
† Middle image: This image, created using X-ray crystallography, shows three positions of mutations in the four-protein complex called DNMT3A. The mutations are disruptions of DNMT3A and are circled in black. The mutations appear different from each other because the proteins are configured differently at each interface. In patients with acute myeloid leukemia, DNMT3A is mutated.
Credit: Norbert Reich, UCSB
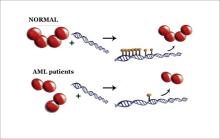
†† Bottom image: In this artist's conception, the four-protein complex called DNMT3A is shown in its normal configuration (top left). The complex reads or "tags" the cell's DNA. In the upper right side of the image, several of these tags are shown on top of the double helix of DNA. The tags control which genes in a cell get turned on and off. In the bottom left image, the complex of four proteins is disrupted. This disruption is caused by the mutation found in patients with acute myeloid leukemia (AML). In the right side of the bottom image, the protein leaves only one tag on the DNA and then moves on.
Credit: Norbert Reich, UCSB
Related Links
Share this article
Related Stories



